Intracellular trafficking, sorting and targeting in health and disease
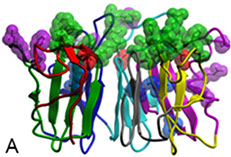
Background. X-linked retinitis pigmentosa type 3 (XlRP3) is a prevalent form of syndromic retinitis pigmentosa (RP), a photoreceptor neurodegenerative disease that exhibits a notable degree of allelic and clinical heterogeneity. This includes an association with partial deafness and increased susceptibility of the upper respiratory tract to infections. The XlRP3 gene encodes the retinitis pigmentosa GTPase regulator (RPGR), whose N-terminal domain shares homology with RCC1 (A), a nuclear guanine-nucleotide exchange factor (GEF) for Ran GTPase. Although RPGR is expressed ubiquitously, its role is selectively critical for photoreceptor neurons (Ba).
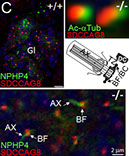
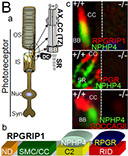
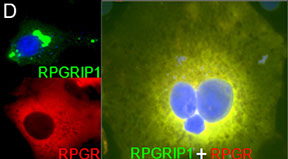
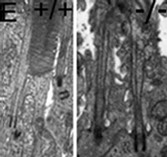
To gain insights into the role of RPGR in photoreceptor function and disease, our laboratory discovered a protein, RPGR-interacting protein-1 (RPGRIP1), which specifically interacts with RPGR and nephrocystin-4 (NPHP4) (Bb). Mutations affecting distinct domains of RPGRIP1 cause its uncoupling from RPGR or NPHP4. NPHP4 causes nephronophthisis type 4, an end-stage renal failure disorder affecting adolescents, or Senior-Løken syndrome, a retinal-renal disease that causes blindness with early onset. Recently, NPHP4 variants have also been linked to cardiac malformations. Among the RPGRIP1 isoforms identified by our laboratory, the RPGRIP1α isoform is selectively expressed in mouse photoreceptor neurons, localizing to their ciliary region (Bc). The lack of RPGRIP1 expression in a mouse model of RPGRIP1 blunts the targeting of RPGR, NPHP4, and other critical ciliary proteins, such as serologically defined colon cancer antigen-8 (SDCCAG8, also called NPHP10/BBS16), to the connecting cilium of photoreceptors, though not affecting various cell types of the kidney (C). Impairment of SDCCAG8 in humans also causes Senior-Løken syndrome, nephronophthisis type 10 (NPHP10), or Bardet-Biedl syndrome (BBS). In cell lines, RPGR prevents the aggregation of RPGRIP1 and promotes its localization to or retrieval from the ER (D). The ultimate physiological effect of RPGRIP1 impairment is the lack of outer segment formation in photoreceptors followed by their rampant degeneration (D). In humans, mutations in RPGRIP1 cause Leber congenital amaurosis (LCA), the leading cause of blindness in children, and RPGRIP1 variants have been associated with several forms of glaucoma (e.g., POAG and NTG).
Current projects. An emerging model supports that RPGRIP1 is crucial for the ER-to-ciliary targeting of its direct partners, RPGR and NPHP4, as well as other higher-order assembly components such as SDCCAG8. However, the dynamic composition of this complex, the biological and functional relationships among its components, and the molecular bases for their cell-context-dependent functions and clinical manifestations, including how these processes contribute to or determine the morphogenesis of subcellular structures (e.g., outer segments), remain elusive. Current studies are focused on refining the RPGRIP1 interactome to investigate routes, components, and mechanisms underlying polarized trafficking and targeting. They also aim to reassess previous models of non-selective bulk flow or cargo-capture transport from the ER.
Moreover, disease mutations affecting all components of the RPGRIP1 interactome will contribute to understanding the biological roles and physiological relevance of these factors in ciliary targeting, subcellular structure morphogenesis, and disease pathogenesis. This overarching goal involves expanding and adapting cell-based assays we developed to probe specific steps of protein routing and examining mouse models of RPGRIP1 and its partners where such processes may be compromised. These approaches will lead to a broader comprehension of intracellular protein trafficking and how its dysfunction contributes to neurodegenerative and other diseases once considered etiologically distinct.
Duke University Medical Center, 2351 Erwin Road, DUEC 3802, Durham, NC 27710, USA