The neuroretina, an experimental system to probe diverse biological
and disease processes
Figure 1. The mammalian neuroretina. Simplified representation of the mammalian retinal network comprising the well-stratified organization of photoreceptor subtypes [rods and red, green and blue cones (C)], second-order neurons [horizontal (H), bipolar (B) and amacrine (A) neurons] and ganglion neurons (G) whose axons extend to the brain, where they synapse to the other neurons. The retinal pigment epithelium (RPE) supports photoreceptor neurons and comprises part of the blood-brain barrier of the retina.
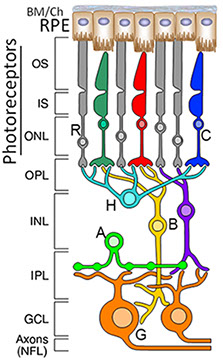
We extensively use, albeit not exclusively, the mammalian neuroretinal network in our studies for several reasons (Figure 1). First, the retina, which lines the inner posterior chamber of the eye, provides a remarkable "window" to study the central nervous system and allied diseases. The neuroretina comprises neurons and glial cells whose cellular and subcellular architectures and functions are extremely diverse, yet relatively well-defined and amenable to experimental examination and manipulation. These features enable the probing of fundamental biological processes, such as long- and short-range signaling, and polarized intracellular trafficking pathways across both diverse and closely related populations of neurons. Second, about one-fifth of the human genes causing Mendelian disease traits contribute to non-syndromic or syndromic vision diseases, often affecting selective retinal neurons, the retinal pigment epithelium (RPE), or both. For example, retinitis pigmentosa (RP) primarily causes the degeneration of rod photoreceptors and the secondary loss of cone photoreceptors, age-related macular degeneration (AMD) leads to the degeneration of RPE and photoreceptor neurons, and glaucoma and optic nerve neuropathies result in impairments and degenerations of ganglion neurons and optic nerve. Furthermore, some of these
and other complex neurological diseases promote the disintegration of neuronal networks through the autonomous and non-autonomous death of selective neurons or supporting cells, by mechanisms that remain ill-defined. Aging and other stresses also act as strong modifiers of the onset, progression, and manifestation of disease. Third, the survival of distinct classes of neurons often depends on the relay of environmental cues to intracellular molecular and subcellular processes that are poorly understood. Dysregulation of these processes and cues often triggers neurodegenerative diseases in selective retinal neurons. Finally, the outcomes of these studies will enable novel insights into signaling and trafficking pathways, and therapeutic approaches to counter impairments of these processes in diseases affecting retinal and other neuronal and non-neuronal systems that share homologous components or pathomechanisms.
Currently, our laboratory is employing two multifunctional and dynamic protein ensembles, Ran-binding protein 2 (RanBP2) and Retinitis Pigmentosa GTPase Regulator-Interacting Protein 1 (RPGRIP1), to investigate signaling and intracellular trafficking pathways, as well as proteostasis in health and disease of selective neurons and other cell types.
Duke University Medical Center, 2351 Erwin Road, DUEC 3802, Durham, NC 27710, USA